Effect of Methionine Loading in Plasma of Healthy Humans | Equine Clinical Research
Clinical Science (1996) 91. 79-86, FMT Loehrer et al.
This excerpt is reprinted by permission of The Biochemical Society and the Medical Research Society, UK. This excerpt may not be reproduced for distribution or sale.
Effect of methionine loading on 5-methyltetrahydrofolate, S-adenosylmethionine and S-adenosylhomocysteine in plasma of healthy humans
Franziska M. T. Loehrer, Walter E. Haefeli, Christian P. Angst, Garry Browne, Greta Frick and Brian Fowler
Metabolic Unit, University Children's Hospitol Basel, and Division of Clinical Pharmacology, University Hospitol Basel, Basel, Switzerland
- Elevated plasma homocysteine concentration, either in the fasting state or after methionine loading, is an independent risk factor for vascular disease in man. Methionine loading has been used to investigate impaired methionine metabolism, especially of the trans-sulphuration pathway, but most studies have focused on changes in homocysteine.
- We investigated the effect of methionine excess on total plasma homocysteine, 5-methyltetrahydrofolate (which is the active form of folate in the remethylation of homocysteine to methionine), S-adenosylmethionine (the first metabolite of methionine) and S-adenosylhomocysteine (the demethylated product of S-adenosylmethionine) over 24h in 12 healthy subjects.
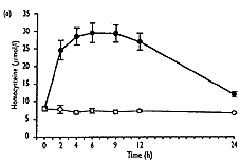
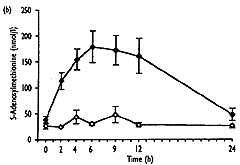
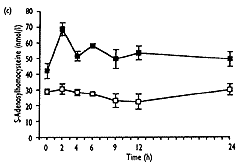
- As well as the expected increase in homocysteine (from 8.0"1.3 to 32.6"10.3 Fmol/l, mean " SD, P<0.001), S-adenosylmethionine showed a significant transient increase (from 37.9 " 25.0 to 240.3 " 109.2 nmol/l, P<0.001), which correlated well with homocysteine (r2=0.92, P<0.001). 5-Methyltetrahydrofolate values decreased significantly (from 23.2 " 7.2 to 13.1"2.9nmol/l, P<0.01), and gradually returned to baseline levels after 24h, No significant change over the time of measurement was found for S-adenosylhomocysteine.
- The sequence of metabolic changes observed in this study strongly suggests that a change in either homocysteine or S-adenosylmethionine may cause a reduction in 5-methyltetrahydrofolate. This must be considered in evaluating the relationship between folate and homocysteine in vascular disease. The metabolic relationships illustrated in this study should be evaluated in the search for pathogenetic mechanisms of mild hyperhomocysteinaemia and vascular disease.
INTRODUCTION
The essential protein amino acid methionine is converted to S-adenosylmethionine by methionine adenosyltransferase (EC 2.5.1.6). S-adenosylmethionine is the methyl donor in many important transmethylation reactions that lead to the formation of the very short-lived S-adenosylhomocysteine [1]. Enzymic hydrolysis yields homocysteine, which can be catabolized via the irreversible transsulphuration pathway, the first step in which is catalysed by the pyridoxal phosphate-dependent enzyme cystathionine b-synthase (CS; EC 4.2.1.22). Alternatively, it can be remethylated to methionine by 5-methytetrahydrofolate--homocysteine methyltransferase (methionine synthase, MS; EC 2.1.1.13), which requires vitamin B12, or by betaine-homocysteine methyl-transferase (EC 2.1.1.5). Regulation of these pathways depends on many factors [2-5], but a vital role of S-adenosylmethionine in the coordinate control of remethylation and trans--sulphuration seems evident from studies in vitro [6,7]. S-Adenosylmethionine acts as an allosteric inhibitor of 5,10-methylenetetrahydrofolate reductase (MTHFR; EC 1.1.1.68), which is crucial for 5-methyltetrahydrofolate synthesis [6] and as an activator of CS [7] at micromolar concentrations.
Disturbances of methionine metabolism are associated with a variety of disease states. For example, inborn errors due to CS, MTHFR and MS deficiencies lead to hyperhomocysteinaemia and severe disease, including ocular, skeletal, neurological and vascular pathology [1,8,9]. Furthermore, elevated plasma homocysteine levels, either in the fasting state or after methionine loading, have been found in a significant proportion of patients with coronary artery [10-13], peripheral arterial occlusive [14,15] or cerebral vascular disease [15-17], pointing to mild hyperhomocysteinaemia as an indep-endent risk factor for arteriosclerotic disease. The exact cause of this is so far unknown, but moderate deficiencies of MTHFR [18-20] and CS [21] and nutritional deficiencies of vitamin B12, and folate [22,23] have been implicated.
Administration of methionine followed by measurement of sulphur amino acids in blood and urine has been used to study homozygous and heterozygous CS deficiency [24-27]. Furthermore, Beers et al. [28] reported post-methionine load increases in homo-cysteine similar to those in obligate heterozygotes for CS deficiency in 36% of 25 patients with peripheral and 28% of 25 patients with cerebrovascular disease. Several studies have confirmed this abnormality in patients with different forms of vascular disease [21,28-30].
It is thought that methionine loading mainly stresses catabolism through homocysteine transsulphuration [31]. However, Clarke et al. [21] showed an inverse relation between vitamin B12, or folate and post-load homocysteine in patients with different forms of vascular disease. On the other hand, in previous studies by Brattstrom et al. [30] and Andersson et al. [32], no correlation between post-load homocysteine and vitamin B12, folate or pyridoxal phosphate was found in patients with vascular disease. Previous studies on the relationship between folate and homocysteine have all used measurement of the total blood concentrations of the vitamin rather than the methyl form, which specifically participates in the remethylation reaction.
Until now methionine loading studies have concentrated on changes in sulphur amino acid levels [33,34], and little is known about the influence of methionine on levels of the numerous other metabolites and cofactors in humans.
In this study the effect of oral methionine on plasma levels of key compounds involved in the transmethylation pathway were studied over 24 h. In particular, and in addition to the often measured homocysteine, we determined the levels of 5-methyltetrahydrofolate, the form of folate active in remethylation of homocysteine, and the intermediate methionine metabolites S-adenosylmethionine and S-adenosylhomocysteine. Additionally, the heat stable activity of MTHFR in lymphocytes was assessed.
 |
 |
 |
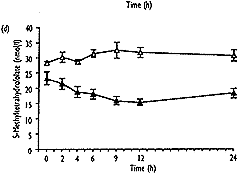 |
| Fig. 2. Concentrations of total homocysteine (a), S-adenosyl-methionine (b), S-adenosylhomocysteine (c) and 5-methyltetra-hydrofolate (d) in plasma after methionine loading in 12 healthy subjects (closed symbols) and without methionine administration in three control subjects (open symbols) over a period of 24 hours. Results are expressed as mean " SEM. |
DISCUSSION
This study set out to investigate the effects of methionine loading on S-adenosylhomocysteine, S-adenosylmethionine and 5-methyltetrahydrofolate and their relation to homocysteine in normal subjects.
That the 12 subjects handled methionine normally was suggested by the finding of normal fasting homocysteine values, which increased to a mean of 32.6" 10.3Fmol/l, which compares with the mean of 35"6Fmol/l found by Mansoor et al. [33]. However, this does not completely exclude the heterozygous state for CS deficiency in all individuals as some overlap of post-methionine homo-cysteine levels between control subjects and obligate heterozygotes has been reported [27]. The variable time to peak values in different subjects in our study emphasizes individual variation in such metabolic studies in humans. It helps to explain the better discrimination of obligate heterozygotes from control subjects observed when several post-load samples were analysed than in simplified tests using a single post-load measurement. Such variable peak level times have important implications both for the studies in vascular disease patients and for investigations of metabolic inter-relationships as in this study.
The major conclusions from this study are that methionine loading in normal human subjects leads to simultaneous increases in homocysteine and S-adenosylmethionine, without significant changes in S-adenosylhomocysteine, and to a later fall of 5-methyltetrahydrofolate. These changes must be consequent to methionine loading and do not simply reflect circadian variations, as demonstrated by the lack of such changes in three control subjects who received no methionine. The increase in S-adenosylmethionine probably reflects liver metabolism of methionine and increases in S-adenosylmethionine concentration as seen in rats injected with a single dose of methionine [44]. This is supported by studies in mammals, which showed that adaptation to an excess of methionine occurs in liver as only the liver-specific isoenzyme of methionine adenosyltransferase can adapt immediately to changes in methionine concentrations [45]. The absence of a change in S-adenosylhomocysteine concentration after methionine loading despite the large increase in homocysteine is surprising. It was shown by Hoffman et al. [46] that in isolated rat liver the administration of homocysteine results in an accumulation of S-adenosylhomocysteine if homocystene is not removed immediately. However, Guttormsen et al. [47] reported no change in S-adenosyl-homocysteine concentration after homocysteine loading, which resulted in similar concentrations of homocysteine to those found after meth ionine loading. If the lack of increase in S-adenosylhomocysteine in plasma does indeed reflect tissue levels, this could be explained simply by a transient increase in homocysteine to concentrations below those required to inhibit S-adenosylhomocysteine hydrolase [46] and by a fully active trans-sulphuration pathway owing to normal enzyme activities in normal subjects.
The decrease in 5-methyltetrahydrofolate found in this study could result from an increase in homocysteine levels, which may result in an enhanced turnover of the remethylation reaction, thereby depleting the co-substrate 5-methyltetrahydrofolate. Alternatively, elevated S-adenosylmethionine could lead to allosteric inhibition of MTHFR, as shown previously in vitro [6], at the concentrations of S-adenosylmethionine obtained in rat liver by injection of similar doses of methionine [44]. It is possible that the two effects may operate in combination. The significant positive correlation between fasting level and both the maximum change and the AUC(t0-24) in 5-methyltetrahydrofolate indicates smaller decreases in 5-methyltetrahydrofolate in subjects with lower baseline values. This suggests that the extent of remethylation in the presence of homocysteine excess may also depend on the availability of 5-methyltetrahydrofolate. The one exceptional subject (VIII) who showed no decrease in 5-methyltetrahydrofolate but the highest post-load level of S-adenosylhomocysteine may reflect the extreme of normal variation. However, these findings could also result from a disturbed remethylation pathway, preventing normal conversion of 5-methyltetra-hydrofolate to methionine, and subsequent stress of the trans-sulphuration pathway reflected by increased S-adenosylhomocysteine. However, this subject exhibited 36% heat-stable activity of MTHFR, which is well above the range of 2.5-4.5 SDs below the mean value, which was defined by Kang et al. [43] as indicating the thermolabile MTHFR mutation.
An additional finding in this study was that the S-adenosylmethionine/S-adenosylhomocyste ratio in fasting human plasma, at 1.2, is lower than that reported for erythrocytes (3.3) [48] and cerebrospinal fluid (6.9) [38], indicating variation of this ratio between different tissues and compartments. Previous workers have shown that this ratio and not S-adenosylmethionine alone is a critical factor in the influence of methyltransferases [49].
The finding that methionine loading leads to decreases in 5-methyltetrahydrofolate subsequent to increases in homocysteine and S-adenosylmethionine indicates that a change in either homocysteine or S-adenosylmethionine may cause reductions in 5-methyltetra-hydrofolate. This must be considered in evaluating the relationship between folate and homocysteine in vascular disease. The metabolic relationships illustrated in this study should be evaluated in the search for pathogenetic mechanisms of mild hyperhomocysteinaemia and vascular disease.
REFERENCES
1. Mudd SH, Levy HL, Skovby F. Disorders of transsulfuration. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGrwHill, 1995: 1279-328.2. Finkelstein JD. Methionine maabolism in mammals. J Nutr Biochem 1990; 1: 228-37.
3. Banerjee R, Matthews RC. Cobalamin-dependent methionine synthease. FASEB J 1990; 4: 1450-9.
4. Selhub I, Miller JW. The pathogenesis of homocysteinemia. Interruption of the coordinate regulation by S-adenosy-lmethionine of the remethylaion and transrulfuration of homocysteine. Am J Clin Nutr 1992; 55: 131-8.
5. Giulidori P, Galli-Kienle M, Catto E, Stramentinoli C. Transmethylation, transsulfuration, and aminopropylation reactions of S- adenosyl-l-methionine in vivo. J Biol Chem 1984; 259: 4205-11.
6. Jencks DA, Matthews RC. Allosteric inhibition of methylenetetrahydrofolate reductase by adenosylmethionine. J Biol Chem 1987; 262:2485-93.
7. Finkelstein JD, Kyle WE, Martin U. Pick AM. Activation of cystathionine synthase by adenosylmethionine and adenosylmethionine. Biochem Biophy Res Commun 1975; 66: 81-7.
8. Rosenblatt DS. Inherited disorders of folate transport and metabolism. In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw-Hill, 1995: 3111-28.
9. Fenton WA. Rosenberg LE. Inherited disorders of cobalamin transport and metabolism. In: Scriver CR, Badet AL, Sly WS, Valle D, eds. The metabolic basis of inherited disease. New York: McGraw-Hill, 1995: 3129-50.
10. Kang SS, Wong PW, Cook HY. Protein-bound homocyt(e)inc: a possible risk factor for coronary artery disease. J Clin Invest 1986; 77: 1482-6.
11. Genest Jr JJ, McNamara JR, Salem DN, Wilson PWE, Schaefer EJ, Malinow MR. Plasma homocyst(e)ine Ievels in men with premature coronary artery disease. J Am Coll Cardiol 1990; 16: lll4-19.
12. Stampfer MJ. Malinow MR, Willett WC, ct al. A prospective study of plarma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA 1992; 268: 877-81.
13. Selhub J, Jacques PE, Bostom AC, et al. Association between plasma homocysteine concentrations and extracranial carotid-artery stenosis. N Engl J Med 1995; 332: 286-91.
14. Malinow MR. Rang SS, Taylor LM, et al. Prevalence of hyperhomocyst(e)inemia in patients with peripheral arterial occlusive disease. Circulation 1989; 79: 1180-8.
15. Taylor Jr LM, DeFrang RD, Harris Jr EJ, Porter JM. The association of elevated plasma homocyst(e)ine with progression of symptomatic peripheral arterial direase. J Vasc Surg 1991; 13: 128-36.
16. Coull BM, Malinow MR. Beamer N, Sexton C, Nordt F, de Garmo P. Elevated plasma homocyst(e)ine concentration as a possible independent risk factor for stroke. Stroke 1990; 11: 572-6.
17. Araki A, Sake Y, Fukushima Y, Matsumoto M, Asada T, Kita T. plasma sulfhydryl-containing amino acids in patients with cerebral infarction and in hypertensive subjects. Atheroscleroris 1989; 79: 139-46.
18. Kang SS, Wong PW, Susmano A, Sora J, Norusis M, Ruggie N. Thermolabile methylenetetrahydrofolate reductase. an inherited risk factor for coronary artery disease. Am J Hum Genet 1991; 48: 536-45.
19. Engbersen AM, Franken DC, Boers CH, Stevens EM, Trijbels EJ, Blom HJ. Thermolabile 5,l0-methylenetetrahydrofolate reductase a a cause of mild hyperhomocysteinemia. Am J Hum Genet 1995; 16: 142-50.
20. Frosst P, Blom HJ. Milos R, et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nature Genet 1995; 10: 111-13.
21. Clarke R, Daly L, Robinson K, et al. Hyperhomocysteinemia: an Independent rirk factor for vascular disease. N Engl J Med 1991; 324: 1149-55.
22. Ubbink JB, Vermaak WJ, van der Merwe A, Becker PJ. Vitamin B12, vitaminB6, and folate nutritional status in men with hyperhomocysteinemea. Am J Clin Nutr 1993; 15: 47-53.
23. Ueland PM, Refusm H. Review article: Plasma homocysteine, a risk factor for vascular disease, plasma levels in health, disease and drug therapy. J Lab Clin Med 1989; 114: 473-501.
24. Laster L, Mudd SH, Finkelstein JD, Irreverre F, Conerly B. Homocystinurea due to cystathionine synthase deficiency: the metabolism of L-methionine. J Clin Invest 965; 44: 1708-20.
25. Mudd SH, Edwerds WA, Loeb PM, Brown MS, Laster L Homocysturnia due to cystathionine synthase defficiency: the effect d Pyridoxine. J Clin Invest 1970; 49: 1762-73.
26. SardharwalaIB, Fowler B, Robins AJ, Komprower GM. Detection of heterozygotes for homocysternia: study of sulphur-containing amino acids in plasma and urine after L-methionine loading. Arch Dis Child 1974; 49: 553-9.
27. Boers GH, Fowler B, Smals AGH, et al. Improved identifiationof heterozygotes for homocysturnia due to cystathionine synthase deficiency by the combinaion of methionine loading and enzyme determinition in cultured fibroblasts. Hum Genet 1985; 69: 164-9..
28. Boers GHJ, Smals AGH, Trijbels FJM, et al. Heterozygosity for homocysternia in premature peripheral and cerebral occlusive arterial disease. N Engl J Med 1985; 313: 709-15.
29. Dudman NP, Wilcken DE, Wang J, Lynch JF, Macey D, Lundberg P. Disordered methionine/homosysteine metabolism in premature vascular disease. Its occurrence, cofactor therapy and entomology. Arterioscler Thromb 1993; 13: 1253-60.
30. Brattstrom L, Israelsson B, Norvving B, et al. Impired homocysteine metabolism in early-onset cerebral and peripheral occlusive arterial disease. Effects of pyridoxine and folic acid treatment. Atherosclerosis 1990; 81: 51-60.
31. Brattstrom L, Israelsson B, Lingard F, Hultberg B. Higher total plasma homocysteine in vitamin B12 deficiency then in heterozygosity for heterocystinurea due to cystathione b-synthase deficiency. Metabolism I988; 37: 175-8.
32. Andersson A, Brattstrom L, Israelsson B, Isaksson A, Hamfelt A, Hultberg B.. Plasma homocysteine before and efter methionine loading with regard to age, gender, end menopausal status. Eur J Clin Invest 1992; 22: 79-87.
33. Mansoor MA, Svardal AM, Schneede ], Ueland PM. Dynamic relation between reduced, oxidized, and protein-bound homocysteine and other thiol components in plasma during methionine loding in healthy men. Clin Chem 1992; 38: 1316-21.
34. Mansoor MA, Bergmark C, Svardal AM, Lonning PE, Ueland PM. Redox status and protein binding of plasma homocysteine and other aminothiols in patients with early onset peripheral vascular disease. Homocysteine and peripheral vascular disease. Arterioscler Thromb Vasc Biol 1995;15: 232-40
38. Weir GG, Molloy AM, Keating JN, et al.. Correlation of the relation of S-adenosyl-L-methionine to S-adenosyl-L-homocysteine in the brain and cerebrospinal fluid of the pig: implications for the determination of this methylation ratio in human brain.. Clin Sci 1992; 82: 93-7.
43. Kang SS, Wong PKW, Zhou J, et al. Thermolabile methylenetetrahydrofolate reductase in patients with coronary artery disease. Metabolism 1988; 37: 611-13.
44. Finkelstein JD, Kyle WE, Harris BJ, Martin JJ. Methionine metabolism in mammals: concentration of metabolites in rat tissues. J Nutr 1982; 112: 1011-18.
45. Baldessarini RJ. Alterations in tissue levels of S-adenosylmethionine. Biochcm Pharmacol 1966; 15: 741-8.
46. Hoffman DR, Marion DW, Cornatzer WE, Duerre JA. S- adenosylmethionine and S-adenosylhomocysteine metabolism in isolated rat liver. Effects of L-methionine, L-homocysteine and adenosine. J Biol Chem 1980; 255:10822-7.
47. Guttormsen AB, Mansoor HA, Fiskerstrand T, Ueland PM. Refsum H. Kinetics of plasma homocysteine in healthy subjects after peroral homocysteine loading. Clin Chem 1993; 39: 1390-7.
48. Perna AF, Ingrosso D, Zappia V, Galletti P, Capasso G, De Santo NG. Enzymatic methyl esterification of erythrocyte membrane proteins is impired in chronic renal failure. Evidence for high Ievels of the natural inhibitor S-adenosylhomocysteine. J Clin Invest 1993; 91: 2497-503.
49. McKeever M, Molloy A, Weir DG, et al. An abnormal methylation ratio induces hypomethylation in vitro in the brain of pig and man, but not in rat. Clin Sci 1995; 88 73-9.