Study: Glutathione in Health And Disease | Equine Clinical Research
The Annals of Pharmacotherapy 1995; 29:1263-73.
Reprinted with permission from The Annals of Pharmacotherapy
www.theannals.com
GLUTATHIONE IN HEALTH AND DISEASE:
PHARMACOTHERAPEUTIC ISSUES
Ben M Lomaestro and Margaret Malone
OBJECTIVE: To review the current research and importance of glutathione (GSH) therapy in health and disease and to provide a basic overview of the widespread use and interest in this compound.
DATA IDENTIFICATION: Articles were obtained via a MEDLINE search of the term glutathione in conjunction with specific disease states mentioned, and via extensive review of references found in articles identified by computer search.
STUDY SELECTION: Emphasis was placed on the most recent research, human research, and in discussing multiple disease states.
DATA EXTRACTION: The literature was reviewed for methodology, quality, and practical aspects of interest to clinical pharmacists.
DATA SYNTHESIS: GSH is a tripeptide of extreme importance as a catalyst, reductant, and reactant. It continues to be investigated in diverse areas such as acute respiratory distress syndrome, toxicology, AIDS, aging, oncology, and liver disease. Despite the widespread clinical interest in GSH, we were not able to identify an in-depth review of this compound in the pharmacy literature.
CONCLUSIONS: The list of potential indications for modulation of GSH is extensive and broad. This review introduces clinicians to what GSH is, its basic chemistry, and some areas of active research.
IN OUR INSTITUTION, we have had recent requests for information on manipulation of glutathione (GSH) concen-trations in patients, and have observed the recent and growing body of literature on this topic. A MEDLINE search on GSH, from 1991 to May 1995, identified more than 8000 published works; however, we were unable to find any review articles in the pharmacy literature. Our purpose was to review the recent literature concerning GSH to introduce clinicians to its complex and diverse functions, its widespread role in maintaining health, and its use in the pharmacotherapy of a variety of diseases.
GSH is a tripeptide of L-glutamate, L-cysteine, and glycine. It is considered to be the most prevalent and most important intracellular nonprotein thiol/sulfhydryl compound in mammalian cells, and the most abundant low-molecular-weight peptide.1-4 It is a cellular reductant, catalyst, and reactant involved in many biologic processes of transport, metabolism, storage, and protection.5 Some of these metabolic functions are presented in Table 1. The mechanisms of GSH activity are both direct and indirect, the latter by maintaining other cellular antioxidants in a functional state.3,6,7 GSH may be especially important for organs that are exposed to exogenous toxins, such as the liver, kidney, lung, and intestines.8 Reduced GSH occurs in millimolar concentrations intracellularly in humans, but in only trace amounts in plasma and most other body fluids. One exception is fluids lining the lower part of the respiratory tract, where it may help to scavenge inhaled toxins and free radicals produced by activated lung phagocytes.9
GSH can be depleted intracellularly either by forming a direct complex with an electrophilic agent (accomplished investigationally by agents such as bromobenzene or diethyl maleate) via inhibition of synthesis, or by subjecting cells to oxidant stress.3 Increasing GSH supply into cells to enhance protection and minimize injury has been proposed in: (1) oxidative injury to the lung from oxygen therapy, cigarette smoke, and/or atmospheric pollutants; (2) oxidative injury to the skin from ultraviolet radiation; (3) injury to the heart and lung from antitumor therapy; and (4) injury to the kidney and small intestine from reperfusion following ischemic events.10 Interventions to increase tissue GSH concentrations have used GSH itself, monoesters that are more completely absorbed, or alternative precursors.
| Table 1. Metabolic Functions of Glutathione |
| DNA synthesis and repair Protein synthesis Prostaglandin synthesis Amino acid transport Metabolism of toxins and carcinogens Enhancement of immune system function Prevention of oxidative cell damage Enzyme activation |
Depletion of intracellular GSH appears to be critical for subsequent alterations in protein thiol and calcium homeostasis.11 GSH depletion and the subsequent low stores of protein thiol result in both calcium release from intracellular stores and inhibition of calcium extrusion, producing a marked increase in cytosolic calcium concentration, which triggers cytotoxicity.11
Glutathione Synthesis
GSH is synthesized from L-glutamate, L-cysteine, and glycine in 2 adenosine triphosphate (ATP)-dependent reactions (Figure 1). The first reaction, catalyzed by gamma-glutamylcysteine synthetase, is effectively rate-limited by GSH biofeedback.1,3,6 The second step involves GSH synthetase, which is not subject to negative feedback by GSH. When GSH is consumed and feedback inhibition is lost, availability of cysteine as a precursor can become the rate-limiting factor.8
Oxidized glutathione (GSSG) is formed in antioxidant reactions that involve GSH, and can accumulate with increased oxidative processing in cells. The ratio of GSSG/ GSH serves as a sensitive index of oxidative stress.12 Because its oxidative functions require GSH to be in its reduced form, GSSG is reduced to regenerate GSH in a reaction catalyzed by GSH reductase that requires reduced nicotinamide adenine dinucleotide phosphate (NADPH) as a hydrogen donor (Figure 2).3,12
Absorption and Distribution of Glutathione
Little is known about the average daily intake of GSH, the amounts of GSH in various food sources, or the importance of dietary GSH in health or disease. The estimated daily intake in humans is about 150 mg of GSH per day.13 Investigations in humans have used 15 mg/kg as an oral bolus to increase the plasma GSH concentration two- to fivefold. The transit of orally administered GSH to tissues is thought to occur via absorption from the intestinal lumen, export from enterocytes into the blood, and uptake from the plasma into cells.10,14 Gastrointestinal transport of GSH appears to be via nonenergy-requiring, sodium-independent, carrier-mediated diffusion.15 The plasma concentration of GSH is low because of its rapid turnover, and more than 80% of plasma GSH is removed by the kidney.16 The serum half-life of GSH after intravenous administration is less than 2 minutes; however, the half-life in epithelial lining fluid is much longer, suggesting independence of respiratory epithelial lining fluid and plasma with regard to GSH metabolism.17
Various epithelial cells, such as enterocytes, alveolar cells, renal proximal tubular cells, endothelial cells, and retinal pigmented epithelial cells, are capable of exogenous GSH uptake, which supports the function of GSH-dependent detoxification systems.10,13 This allows GSH concentrations to be maintained better than by synthesis alone. Increasing plasma GSH concentrations by oral administration has been shown to increase the availability of GSH for transport into these tissues.13 This provides the basis for augmenting GSH concentrations against a wide variety of pathophysiologic states, including hepatic dysfunction or cirrhosis, or conditions affecting the epithelial cells, which can use exogenous GSH for protection.
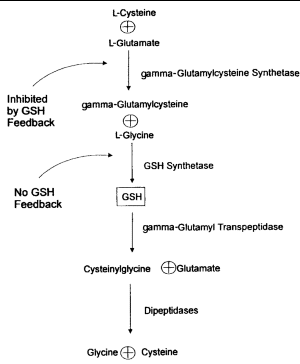 |
| Figure 1. Synthesis and breakdown of glutathione (GSH). |
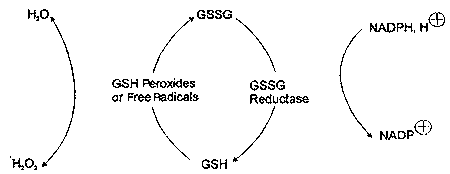 |
| Figure 2. Relationship of oxidized (GSSG) to reduced (GSH) glutathione. |
Most cells, in contrast to epithelial cells, do not have a direct transport capacity for intact GSH.3 Substrates for GSH synthesis are provided either by transport of amino acids into the cells or by transpeptidase activity at the cell surface, which is responsible for salvaging amino acids from circulating GSH for reuse in the intracellular resynthesis of GSH. The cellular concentration of GSH, therefore, is regulated by a complex process of precursor amino acid transport across cell membranes, intracellular synthesizing enzymes, feedback regulation, and intracellular GSH complexing via conjugation of GSH with a variety of electrophilic compounds through GSH transferase reactions.
Although GSH is synthesized from precursors in virtually all cells, the liver is the main source of plasma GSH.18 In animal models hepatic concentrations of GSH change diurnally, with the highest concentration at 1000 hours and the lowest at 1800 hours.18,19 The plasma concentration of GSH is a function of hepatic synthesis, oxidation-reduction reactions, extrahepatic uptake and degradation, and GSH absorption.l3 In the liver, distribution of GSH is heterogeneous, which may play a significant role in susceptibility to acute hepatotoxicity from various toxic compounds such as acetaminophen metabolites, redox cycling compounds, peroxides, and others.
Most GSH clearance from plasma occurs in the kidneys and the lungs.1,8,20,21 In the kidneys, exposure to toxins and the requirement for detoxification by GSH are high. Steady-state intracellular GSH concentrations vary in different parts of the kidneys, which also may determine the localization of injury by toxins.8
Role of Cysteine Homeostasis in Glutathione Metabolism
Cysteine provides the reactive thiol group, which is key to the function of GSH.8 It is required for hepatic GSH synthesis and may be derived from methionine, which serves as a major source of cysteine, via the trans-sulfuration pathway of the liver.18 Cysteine also inhibits hepatic GSH efflux. It cannot be transported in/from plasma or stored within the cell as cysteine, as it would rapidly autooxidize to cystine, producing potentially dangerous oxygen radicals.8 This toxic autooxidation is avoided by storing almost all nonprotein-bound cysteine as GSH.1,8 Cysteine occurs in plasma in the following forms: the solitary amino acid with its free thiol group (free cysteine), a disulfide between 2 molecules of cysteine (cystine), and a mixed disulfide with cysteinyl residues of albumin or other plasma proteins (protein-bound cysteine).22
The kidneys use cystine as a source of cysteine, and they clear and break significant quantities of plasma GSH into component amino acids.8 Therefore, the kidneys are a major source of plasma cysteme that can be used by the liver for resynthesis of GSH.
Major Functions of Glutathione
The major functions of GSH can be explained by its role in detoxification, redox reactions, and the storage and transport of cysteine. Because a major physiologic function of GSH is to provide cells with a reducing environment and to destroy the reactive oxygen compounds and free radicals formed in metabolism, organs that have low concentrations of other antioxidants (such as catalase and superoxide dismutase) are thought to be more dependent on GSH for detoxification of reactive oxygen species than are organs that have alternative antioxidants.20
DETOXIFICATION
Either by spontaneous conjugation or by reduction, GSH provides the bulk of available sulfhydryl groups for binding and detoxification of reactive endogenous and exogenous compounds such as peroxides and electrophiles.2,23,24 GSH peroxidase is an enzyme present in tissues,25 which converts peroxides, using GSH as a substrate, into less harmful fatty acids, water, and GSH disulfide.26 These reductive reactions generate the oxidized form of GSH, GSH disulfide (GSSG).24 Under normal circumstances, to preserve high inuacellular concentrations of free GSH, GSSG is reduced rapidly back to GSH by the NADPH-dependent GSSG reductase. In this way a high GSH/GSSG ratio is maintained within the cells.24 However, GSSG is actively excreted from the cell when its intracellular concentration exceeds the reductive capacity of the cell, as in conditions of oxidative stress. The GSH/GSSG ratio is hypothesized to affect the redox balance of protein thiols, and to regulate the activity of certain enzymes by disulfide exchange reactions.
The capacity for GSH synthesis is insufficient to maintain GSH concentrations when tissues are exposed to certain drugs or their metabolites (e.g., acetaminophen), redox cycling compounds (e.g., menadione), peroxides (e.g., tertbutyl hydroperoxide), X-rays, or ultraviolet radiation.13 Its depletion has been associated with enhanced toxicity of many compounds that cause increased morbidity or death.2 For example, hepatotoxicity of acetaminophen is caused by the production of a highly reactive intermediate oxygen metabolite (N-acetyl-p-benzoquinoneimine). This metabolite covalently bonds to tissue macromolecules, resulting in tissue injury and cell death (Figure 3).25,27,28 Ito et al.25 have shown that inhibition of the GSH redox cycle enhances acetaminophen cytotoxicity in cultured rat hepatocytes. Excessive amounts of acetaminophen can overwhelm the capacity of GSH for conjugation and lead to toxicity.27 Also, fasting (by shunting acetaminophen metabolism away from glucuronidation or by depletion of intracellular GSH) and chronic ethanol use (via intracellular GSH depletion) can increase susceptibility to acetaminophen hepatotoxicity.28
GSH also may activate parent compounds by their metabolic conversion to active intermediates. For example, the metabolism of nitroglycerin requires interaction with thiols such as cysteine and GSH for transformation to vasoactive metabolites such as nitrosothiols or nitric oxide.29 In addition to activation of nitroglycerin via interaction with thiol compounds such as cysteine and GSH, dilation of small vessels only responsive to nitroglycerin in the presence of exogenous cysteine has been proposed. Finally, N-acetyl-cysteine (NAC) has been shown to partially prevent tolerance development to nitrate-induced antianginal effects.
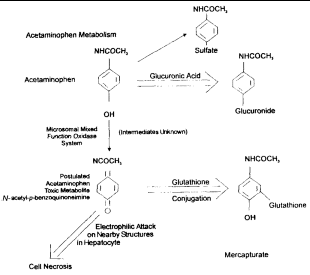 |
| Figure 3. Acetaminophen metabolism and glutathione. |
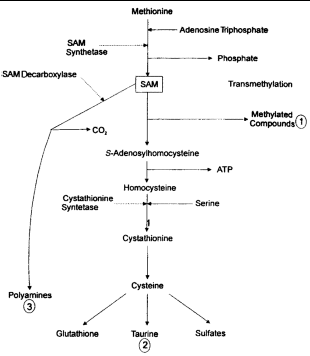 |
| Figure 4. Hepatic synthesis of glutathione from S-adenosyl-L-methionine (SAM) (adapted from references 5, 23 and 41). |
GLUTATHIONE AS AN ANTIOXIDANT
Oxygen radical stress occurs in all aerobic organisms as a result of aerobic metabolism, with intermediates such as superoxide and hydrogen peroxide that lead to further production of oxygen radicals, which can cause lipid peroxidation and disrupt metabolic processes.8 Antioxidant defenses are not completely efficient, and increased free-radical formation (oxidative stress) is likely to increase the damage.9 The GSH redox cycle (the balance between GSH and GSSG) has been shown to be more effective than catalase in hydrogen peroxide detoxification in endothelial cell cultures.12
GSH serves as a substrate for 2 antioxidant enzymes: GSH peroxidase and phospholipid hydroperoxide GSH peroxi-dase.12 Glutathione-S-transferase is also a glutathione- dependent enzyme, mainly involved in xenobiotic and lipid peroxide detoxification.12 Although GSH exerts antioxidant properties through antioxidant enzymes, it also provides protection against oxidative stress by nonenzymatic free radical scavenging.12 Reduced GSH is capable of directly scavenging radicals and peroxides by being oxidized to either GSSG or to a mixed disulfide, thereby preventing cell membrane lipid peroxidation and its subsequent deleterious effects on cellular functions.4
Various oxygen radical stresses have been shown to result in GSSG formation and short-term depletion of GSH.3 It has been postulated that the oxidation-reduction status of GSH may act as a third messenger in either enhancing or diminishing the activities of a number of biologic processes, such as enzyme catalysis, protein synthesis, and receptor binding.30
Relationship of S-adenosyl-L-Methionine and Glutathione
The metabolism of GSH and S-adenosyl-L-methionine (SAM) are closely linked, as the liver forms GSH as a product of SAM metabolism (Figure 4). The liver uses as much as 70% of dietary methionine, and most is converted to SAM.31,32 SAM is an important metabolic substrate and is involved in the initiation of 3 major pathways: (1) transmethylation for the synthesis of various proteins and lipids, among them phospholipids for cell membranes; (2) trans-sulfuration to form GSH and sulfated compounds via homocysteine and cysteine; and (3) aminopropylation for polyamine synthesis.33
Glutathione in Disease
LIVER DISEASE
Decreased SAM use by the liver can result in reduced GSH availability and potential hepatotoxicity.34 Patients with cirrhosis of the liver have hypermethioninemia and a block of the trans-sulfuration pathway leading to delayed methionine clearance and decreased concentration of methionine endproducts (including GSH) following a methionine load.32,33,35 The lack of accumulation of intermediary metabolites suggests that the defect is located in the initial step of methionine transformation regulated by SAM synthe-tase.33,34 Because GSH is vital in detoxification and cell physiology, its depletion has been speculated to represent an important contributing factor of liver injury, and enhanced morbidity in patients with liver injury.2
Chawla et al.22 measured the concentrations of cysteine, GSH, and taurine in 14 healthy subjects and 10 patients with cirrhosis fed either mixed food, nasogastric hyperalimentation with Vivonex (Sandoz Nutrition, Minneapolis, MN), or FreAmine III (McGraw, Irvine, CA) intravenous hyper-alimentation. Vivonex and FreAmine III do not contain cysteine. Subnormal plasma concentrations of GSH and cysteine were observed in patients with cirrhosis independent of their diet. The data support the hypothesis of an acquired dysfunction in the hepatic trans-sulfuration pathway, rather than a change in bioavailability. Because most plasma GSH originates in hepatocytes, the authors hypothesized that decreased plasma GSH also could signify intracellular depletion. This would potentially impair the ability of the hepatocyte to maintain normal redox potential, destroy peroxides and free radicals, and detoxify drugs.22
Loguercio et al.16 evaluated the plasma and erythrocyte GSH and cysteine concentrations of patients with liver cirrhosis with respect to alcoholic or nonalcoholic etiology, severity of liver disease, and nutritional status. The data demonstrated a four- to eightfold decrease in plasma GSH content in 48 cirrhotic patients versus a control group of 18 healthy volunteers. This decrease was irrespective of cirrhosis etiology and thought to reflect diminished hepatic GSH synthesis. A significant decrease in cysteine in severe cases of cirrhosis also was observed. It is known that plasma concentrations of cysteine are affected by diet composition and fasting. However, cysteine also is provided by methionine through the trans-sulfuration pathway, which if impaired could contribute to the depression of plasma cysteine concentration. This investigation found no correlation between plasma cysteine and nutritional status, pointing to impairment of trans-sulfuration as the major cause.
Altomare et al.2 measured reduced and oxidized hepatic GSH concentrations during alcoholic and nonalcoholic liver injury in 35 chronic alcoholics, 20 nonalcoholic patients with liver disease (chronic active hepatitis, chronic persisting hepatitis, steatosis, cirrhosis), and 15 control patients (admitted for uncomplicated abdominal procedures). Decreased GSH concentrations were noted in patients with alcoholic (2.55 ± 0.1 mol/g of liver) and nonalcoholic liver diseases (2.77 ± 0.1 mol/g of liver) compared with controls (4.14 ± 0.1 mol/g of liver) that were speculated to be a contributing factor in liver injury and could enhance the risk of hepatic toxicity from a variety of toxic agents. The GSSG concentration was also significantly higher in alcoholics (8.2 ± 0.3% of total) and nonalcoholics (8.5 ± 0.8% of total) than in control subjects (4.4 ± 0.2% of total), reflecting an abnormal balance between GSH and GSSG in these patients. The investigators concluded that decreased hepatic GSH concentrations in patients with liver disease may represent a contributing factor of liver injury susceptibility and toxicity risk in these patients. Furthermore, excess GSSG is postulated to result in alteration of a variety of cell functions, including enzyme function, protein synthesis, cell integrity, microtubular function, transport processes, and release mechanisms.2
Data suggest that plasma and hepatic GSH also are decreased in patients with acute viral hepatitis, or chronic liver injury from a variety of causes such as chronic hepatitis, nonalcoholic liver cirrhosis, and alcoholic liver disease.36,37 The etiology again is thought to include altered SAM metabolism in patients with chronic liver disease regardless of the stage or cause of the disorder.23
Seifert et al.38 found a correlation between liver disease in chronic alcoholics and a subsequent GSH depletion state. Chronic alcoholic patients between the ages of 21 and 65 years were investigated for predisposition to acetaminophen hepatotoxicity caused by increased activity of the cytochrome P450 system and decreased hepatic GSH concentrations. Venous blood was analyzed for liver profile, prothrombin time, GSH, and GSSG. Acetaminophen use and diet appeared to have no effect on plasma GSH concentrations. Total GSH in patients with normal gamma-glutamyl transferase (GGT) did not differ significantly from that in healthy volunteers. Acetaminophen toxicity was not observed. However, patients with an elevated GGT (102 IU/L) had significantly lower plasma total GSH concentrations (2.38 ± 2.42 mol/L vs. 5.81 ± 4.11 mol/L, p < 0.0001). These data suggest that in chronic alcoholics with liver disease, GSH deficiency exists, which may predispose to further liver toxicity caused by resultant inadequate defense mechanisms.
ROLE OF GLUTATHIONE IN THE IMMUNE SYSTEM: AIDS DATA
Many of the pathologic aspects of disease in patients with AIDS are not caused directly by the HIV infection, but are secondary effects caused by the host response to infection.39 One of the more important aspects of this disease is the chronic inflammatory and oxidative stresses that accompany the infection, which eventually contribute to the loss of CD4 (helper) T-cells, increased opportunistic infections, immuno-deficiency in general, wasting disease, and death. Further-more, HIV infection is characterized by a systemic GSH deficiency, which has been postulated to increase viral replication and to increase production of oxidants from inflammatory cells possibly contributing to immune system dysfunction.17
The consequences of GSH depletion in AIDS and the role of thiol replacement therapy have been described.39-44 The progression of HIV infection from its early asymptomatic stage to active late-stage AIDS is thought to begin with the production of inflammatory cytokines that stimulate the production of the latent virus.42 Adjuvant therapy with GSH replacement in patients infected with HIV could offer several potential benefits. For example, stimulation of viral replication by inflammatory cytokines is optimal at decreased glutathione concentrations.39,40 GSH and NAC effectively block the cytokine-induced production of virus and may extend the latency period of early infection.39,40 Restoration of depleted GSH concentrations in T-cells may be critical for restoration of leukocyte function.39
Intracellular GSH has been shown to play an important role in aspects of T-cell function, including the binding, internalization, and degradation of interleukin-2, as well as DNA synthesis.4 In particular, GSH and sulfhydryl compounds are known to augment the activation of cytotoxic T-cells in mixed lymphocyte cultures, T-cell proliferation in response to mitogens, and the differentiation of T and B lymphocytes.45 In vivo administration of GSH has been demonstrated to enhance the activation of cytotoxic T-cells, but depletion of GSH intracellularly inhibited the activation of lymphocytes, increased susceptibilities of human lymphoid cells to radiation, and suppressed cell-mediated cytotoxic functions, suggesting that intracellular GSH can modulate the function of immune cells. The current hypothesis is that because adequate concentrations of GSH are required for proper lymphocyte function, a deficiency in GSH may contribute to the immunodeficiency seen in the later stages of HIV infection.41 In addition, the action of inflammatory cytokines may mediate cachexia and the wasting that accompanies late stages of AIDS, which also may be alleviated by GSH replacement.40,42
Holroyd et al.17 investigated the effect of administering reduced GSH 600 mg via aerosolization twice daily to 14 HIV-seropositive individuals. GSH concentrations in the lung epithelial lining fluid were compared before and at 1, 2, and 3 hours after administration. Total GSH concentrations increased and remained in the normal range for the 3 hour post-treatment studied. A striking increase in oxidized GSH (GSSG increased from 5% at baseline to about 40% 3 h after treatment) was noted, possibly reflecting the potential value of GSH as an antioxidant providing protection in the lung tissue.
NAC is a cysteine prodrug capable of maintaining intra-cellular thiol concentrations and replenishing GSH in deficiency states. It blocks HIV expression in acute and chronic infection models, and HIV replication in normal peripheral blood mononuclear cells.40 The administration of NAC has been mentioned as a "new approach to anti-HIV therapy," which differs from existing antiviral drugs in that it inhibits host-mediated stimulation of viral replication arising in normal immune responses, and may hence extend latency. In vitro, NAC has been shown to block cytokine-stimulated HIV replication in an acutely infected T-cell line and in acutely infected peripheral blood mononuclear cells from healthy individuals.42
PULMONARY DISEASE
GSH deficiency has been proposed to have a role in the pathophysiology of a number of lung diseases, including chronic obstructive pulmonary disease,46 acute respiratory distress syndrome (ARDS),47-49 neonatal lung damage,50 and asthma.51 The lung is particularly at risk from oxidative damage as it is exposed to oxygen, oxygen radicals produced by alveolar macrophages, inhaled environmental and blood-borne toxins, including cigarette smoke and atmo-spheric pollutants.8,10 Free radical induced toxicity is worsened by concomitant GSH deficiency, and free radical production further depletes GSH through use.52 GSH present in the epithelial lining fluid of the lower respiratory tract may be the first line of defense against oxidant stress.8 In idiopathic pulmonary fibrosis, for example, GSH concentrations are only 25% of normal values in the epithelial lining fluid and may be involved in the underlying pathophysiology of the disease.8
Neonates. Pulmonary GSH concentrations have been found to be lower in premature infants with a lower gestational age (25 vs. 39.8 wk average gestational age).53 Theories to explain why pulmonary GSH could be depleted in some infants include decreased cord cysteine concentrations, seen in earlier gestational age, that may impair GSH synthesis as intracellular GSH depends on the availability of cysteine. This hypothesis was investigated by Grigg et al.50 The concentration of GSH in bronchoalveolar lavage fluid was measured on the first day of life in intubated infants born at less than 35 weeks gestation irrespective of initial respiratory status. Lower concentrations of GSH were observed in 7 infants who subsequently developed chronic lung disease when compared with 27 infants who did not require oxygen supplementation at 36 weeks postconceptional age. This investigation provided preliminary evidence that a low lung epithelial lining fluid concentration of GSH on the first day of life is associated with an increased risk of chronic lung disease. However, a method of increasing GSH concentrations in this population was not discussed.
Asthma. Airway inflammation in asthma also is associated with the increased generation of reactive oxygen species and pathophysiologic changes.51 In a comparison of 10 adults with mild asthma versus 17 healthy volunteers, concentrations of GSH in bronchoalveolar lavage fluid were increased in the subjects with mild asthma. The mean bronchial GSH concentrations were 13.0 nM/mg of protein in healthy volunteers versus 23.9 nM/mg of protein in mild asthmatics. The mean alveolar GSH concentration was 23.3 nM/mg of protein in volunteers versus 36.5 nM/mg of protein in mild asthmatics. The authors hypothesized that an increasing GSH concentration represents an adaptive mechanism to increase antioxidant defenses, which results in fewer symptoms, reduced airway reactivity, and milder disease in patients with asthma.
Acute Respiratory Distress Syndrome. GSH is detected in high concentrations in the extracellular epithelial lining fluid of the lower respiratory tract of healthy subjects, and could act as a first-time scavenger of toxic oxygen intermediates and protect against lung cell damage and injury.l7 Patients with ARDS and sepsis have a GSH deficiency in the lower respiratory tract extracellular epithelial lining fluid, which could favor oxidative stress and subsequent damage.47,48 Also, greater percentage of total GSH exists in the oxidized form in patients with ARDS than in healthy subjects, possibly indicating increased oxidant stress in the lower respiratory tract of ARDS patients.49 The mechanism for GSH deficiency is thought not to be dilutional (as occurs in cardiogenic pulmonary edema), but representing depletion of tissue stores.
GSH depletion in plasma and granulocytes in ARDS is reversible by NAC therapy. Suter et al.47 improved systemic oxygenation and reduced the need for ventilatory support in 32 patients with acute lung injury caused by various underlying diseases, with NAC 40 mg/kg/d administered as a continuous infusion over the first 3 days after admission to the intensive care unit. Patients in the NAC group also had a shorter median stay in the intensive care unit than did those in the placebo group (median 7 vs. 10 d, respectively).
ONCOLOGY
By acting as an antioxidant and by virtue of binding to cellular mutagens, GSH has the ability to react with peroxides and several electrophiles, including carcinogenic epoxide metabolites.7,54 GSH has been shown to directly modulate proliferation of highly purified T-cells, suggesting that GSH is essential for steps closely involved with DNA synthesis. Depressed intracellular GSH in the liver and in mammary tissue has been shown to promote carcinogen binding to DNA.54 Oral glutamine, for example, has been proposed to have a useful role in increasing host GSH concentrations in the gut, liver, lung, kidneys, heart, and muscle after exposure to radiation or chemotherapy without enhancing tumor growth.55
Flagg et al.7 investigated the association between dietary GSH intake and the risk of oral and pharyngeal cancer in an epidemiologic study of 1830 participants. In this case-control study, the investigators noted an inverse relationship between dietary intake of GSH and the risk of oral cancer, but only in a cohort consuming GSH mostly from raw fruit and vegetables (rather than meat or cooked vegetables, for example). However, the possibility that GSH intake from fruit and vegetables might be protective against oral cancer risk could not be distinguished from the more general benefit of consuming raw fruits and vegetables, such as increased ingestion of fiber. The investigators hypothesized anticarcinogenic protective mechanisms of GSH to include its direct antioxidant function, indirect maintenance of other antioxidants, possible mediation of DNA synthesis and repair, and the ability to bind with cellular mutagens.3
Resistance to chemotherapy caused by detoxification by GSH has been postulated,56 suggesting that there may be possible benefits of selectively decreasing intracellular GSH in tumors to enhance the therapeutic effect of chemotherapy. Direct bonding to the sulfhydryl group of GSH in the cytoplasm has been shown to inactivate cisplatin, for example. Further, GSH depletion can inhibit DNA repair in bladder carcinoma cell lines after cisplatin or doxorubicin damage. Limited data suggest that GSH and its related enzymes enhance drug resistance, varying with the characteristics of the tumor and the chemotherapy chosen. The use of buthionine sulfoximine, which inhibits gamma-glutamylcysteine synthetase, has been investigated in animals as a method to decrease GSH.3 With elucidation of its mechanism, incidence, and impact on resistance to chemotherapy, interventions to decrease target concentrations of GSH may become part of future oncologic therapeutic regimens.
PREECLAMPSIA
Walsh and Wang26 determined that GSH peroxidase activity is significantly lower, and lipid peroxides and thromboxane significantly higher, in preeclamptic placentas than in healthy placentas because of an unknown mechanism. They postulated that peroxides stimulate prostaglandin H (PGH) synthase, leading to the generation of oxygen radicals and increased formation of both thromboxane and lipid peroxides. They proposed that PGH synthase converts arachidonic acid into prostaglandin G2 and prostaglandin H2. Thromboxane synthase then converts prostaglandin H2 into thromboxane A2. When PGH synthase is activated, the peroxidase function of the enzyme generates oxygen radicals that could interact with polyunsaturated fatty acids in the placenta to form lipid peroxides. GSH peroxidase inactivates peroxides, using GSH as its cofactor to convert lipid peroxides to less harmful hydroxylated fatty acids, water, and GSH disulfide. If GSH peroxidase activity is deficient, lipid peroxidation could increase in the tissues, leading to increased stimulation of PGH synthase, increased thromboxane production, and further increase lipid peroxides. Deficiency of this enzyme would likely increase morbidity in preeclampsia.
PARKINSON'S DISEASE
The brains of patients with Parkinson's disease exhibited a reduction of GSH, selective to the substantia nigra (SN). This did not appear to be related to drug therapy. It was postulated to be of significance in the pathogenesis of this disease via production of oxidative damage.57,58 These patients had an increased concentration of the GSH degradative enzyme gamma-glutamyltranspeptidase in the SN, and a normal concentration of the synthetic enzyme gamma-glutamyl-cysteine synthetase.57 GSH depletion occurred without a change in GSSG, suggesting efflux of GSH out of the glia, perhaps with additional increased conversion of GSH to GSSG in response to increased hydrogen peroxide formation. At this time it is unclear whether free radical involvement in Parkinson's disease is a primary or secondary event in nigral cell death or whether it occurs early or late in the disease process.58
SEPSIS
Henderson and Hayes59 reviewed the existing data on NAC as an antioxidant in patients with severe sepsis. They noted that GSH is depleted in severe sepsis, possibly in the defense against active radicals generated by inflammatory cells during systemic inflammatory response syndrome. These inves-tigators are experimenting with a regimen of NAC 150 mg/kg in dextrose 5% infused intravenously over 30 minutes followed by 15 mg/kg/h for 4 days as an antioxidant active radical scavenger. Their hypothesis is that NAC may improve renal function, reduce fluid requirements, and lower tissue edema.
Spies et al.60 reported a 2-year investigation of 58 patients with septic shock randomized to receive NAC 150 mg/kg iv over 15 minutes followed by 12.5 mg/h over 90 minutes. Using a 10% or more increase in whole body oxygen con-sumption as their endpoint, 13 patients who received NAC were considered responders, and 16 were considered nonresponders. None of the patients who received placebo (n = 29) exhibited a 10% increase in whole body oxygen consumption. In summary, NAC transiently improved tissue oxygenation in about half of the patients with septic shock to whom it was administered.
MYOCARDIAL ISCHEMIA AND REPERFUSION INJURY
During myocardial ischemia and reperfusion, both myocardial GSH and the GSH/GSSG ratio within the ischemic tissues are reduced, and the extent of the myocardial injury is inversely dependent on the myocardial GSH content.61-63 Treatment with gamma-glutamylcysteine ethyl ester can increase intracellular reduced GSH concentrations, and has been shown to result in a dose-dependent reduction in infarct size in a canine model of occlusion-reperfusion.61 However, an investigation of coronary artery occlusion in 23 mongrel dogs administered NAC 30 minutes before and continued until 3 hours after reperfusion failed to observe an increase in total GSH or GSH peroxidase activity at the biopsied ischemic zone, or a decrease in myocyte death.64
RENAL DYSFUNCTION AND NEPHROTOXICITY
We have alluded to the potential for decreases in GSH concentration to impair GSH conjugation and modify the metabolic fate of many compounds. GSH deficiency may contribute to the nephrotoxicity of ischemic events and drug toxicity. This type of toxicity may be exhibited by cyclosporine. Although the exact mechanism of cyclosporine nephrotoxicity remains unknown, its administration has been associated with in vivo reduction of GSH concentrations in the livers and kidneys of rats, which may be related to adverse effects of this immunosuppressive agent.65,66 Cyclosporine has peroxidative properties, induces lipid peroxidation in renal microsomes, and may lead to inactivation of microsomal glucose-6-phosphate activity and toxicity.66,67 Therefore, contribution to cyclosporine nephro- and hepatotoxicity has been postulated to be caused by its generation of free radicals and depletion of GSH. Investigators also have hypothesized that cyclosporine can modify resistance to chemotherapy by augmenting the cytotoxic effect of drugs through inducing a GSH deficiency.65
The ability of GSH administration to prevent nephrotoxicity from renal ischemia, and consequent production of oxygen free radicals, has been investigated. A total of 200 mg/kg of GSH (available in Japan as Tathion, Yamanouchi Pharmaceutical, Tokyo, Japan) was administered intra-venously to 10 patients before cardiopulmonary bypass surgery, and the influence on postoperative renal dysfunction was compared with 9 other patients undergoing the same procedure without GSH administration.16 Administration of GSH resulted in a significantly higher urine volume (approximately 20%) on the first and second postoperative days, with a trend to lower blood urea nitrogen and plasma creatinine concentrations in the GSH group that did not reach statistical significance. Also, the mean arterial pressure and systemic vascular resistance index were lower than those in the control group. The investigators concluded that administration of exogenous GSH had a beneficial effect on renal function by virtue of its antioxidant properties, and possibly by a vasodilator action to increase the glomerular filtration rate as well.
Role of Glutathione in Aging
Although higher GSH concentrations have been associated with good health, the significance of low GSH status in the elderly is inferred from limited data.68,69 Investigators have noted lower GSH concentrations to be associated with the combination of advanced age and increased risk of chronic diseases such as chronic renal failure, malignant disorders, diabetes, alcoholism, Parkinson's disease, and cataract formation.
Julius et al.68 measured GSH concentrations in 33 people between 60 and 79 years of age, and related the data to health, the number of illnesses, and other risk factors for chronic disease. There appeared to be a direct relationship between higher GSH concentrations and increasing age with good health. The association of GSH with good health was positive and independent of age (i.e., volunteers with higher GSH concentrations were healthier than age-matched volunteers with lower GSH concentrations). People with chronic diseases had lower mean GSH concentrations than those who were free of disease.
Lang et al.69 compared GSH blood concentrations in healthy young and healthy elderly subjects and found that the reference group of 20- to 39-year-old subjects (n = 40) had a GSH concentration 17% higher on average than the 60- to 79-year-old cohort of 60 subjects (mean ± SD, 547 ± 53.5 g/1010 vs. 452 ± 86.8 g/1010 erythrocytes, respectively). Caution has been advised in interpreting these small studies to define the association of GSH with aging.70 Further large-scale studies are needed to prove that low relative GSH concentration is an overall risk factor for morbidity among the elderly.
Pharmacotherapeutic Interventions to Increase Glutathione Concentrations
GLUTATHIONE AND RELATED AGENTS
GSH is present to the greatest extent in fruits, vegetables, and meats.71 Agents such as 1-cyano-2-hydroxy-3-butene, present in cabbage, brussels sprouts, broccoli, and cauliflower, have raised GSH concentrations severalfold in animal models.72 The results of studies to date of GSH absorption have been conflicting. Several investigators have shown that orally administered GSH increases plasma concentrations of reduced and protein-bound GSH through intestinal absorption in animal models10,13,15 and humans.73
GSH is not commercially available as an oral or injectable product in the US because of pharmaceutical problems, including poor oral bioavailability and a short halflife (2 min) with intravenous administration. Investigators have used bulk quantities of GSH purchased from various chemical companies. For these reasons, precursors of GSH have been investigated. In mice pretreated with buthionine sulfoxime to inhibit GSH synthesis and induce a deficient state, oral administration of GSH resulted in statistically significant increases in GSH concentrations in kidney, heart, lung, brain, small intestine, and skin, but not in the liver. Administration of the equivalent amount of the constituent amino acids to GSH-deficient mice resulted in little change in GSH concentration in all tissues studied. Mice not pretreated with buthionine sulfoximine, but administered GSH, experienced an increase in plasma GSH, but not tissue GSH.10 In humans, oral doses of GSH 15 mg/kg increased plasma GSH 1.5- to 10-fold over the basal concentration in 4 of 5 volunteers tested.73 The maximum concentration of plasma GSH generally occurred 1 hour after GSH administration. Equivalent amounts of amino acid constituents of GSH failed to increase plasma GSH concentrations. These data suggest that oral GSH can replete GSH concentrations in several tissues following GSH depletion, such as after toxicologic or pathologic conditions that alter GSH homeostasis.10
It has been speculated that because oral administration of GSH leads to its inactivation by peptidases, it should not be possible to significantly increase plasma GSH concentrations with oral administration.74 Witschi et al.74 investigated the bioavailability of a single dose of oral GSH in 7 healthy volunteers who had fasted. Their data showed a nonsignificant increase in plasma GSH after doses as high as 3.0 g, suggesting negligible systemic availability of oral GSH in humans. Cook and Sherlock75 also were not able to demonstrate a benefit from oral GSH (100 mg tid for 28 d) in 10 patients with hepatic cirrhosis of various etiology. Analysis included measurement of biochemical parameters and an assessment of sense of well-being compared with 10 other patients with cirrhosis not administered GSH. In a separate arm of investigation, another 12 patients with cirrhosis received GSH 200 mg/d im without producing any difference in biochemical parameters. This latter GSH group did note an increased sense of well-being versus the control group from the oral GSH investigation.
Evidence suggests that cells export GSH, but evidence that GSH is transported into cells to any appreciable extent is conflicting.5,10,73 Therefore, methyl, ethyl, and isopropyl esters of GSH in which the glycine carboxyl group is esterified have been used and are orally bioavailable via rapid intracellular deesterification with effective transport and hydrolysis intracellularly.5,61 As an example, orally administered GSH ethyl ester is able to significantly raise GSH concentrations in the liver, kidney, spleen, pancreas, heart, and lung of the mouse, and in human red blood cells, skin fibroblasts, and several lymphoid cell lines.5 Various esters and amides of GSH are transported into cells, but some have toxicity caused by cleavage products such as methanol, ammonia, and alcohols.5 None is commercially available in the US at this time.
Gamma-glutamylcysteine ethyl ester was studied recently as a GSH precursor used for myocardial protection in dogs with ischemia and reperfusion damage.61 When administered as 3 or 10 mg/kg intravenously immediately before reperfusion in a canine coronary occlusion-reperfusion model of myocardial infarction, a significant dose-dependent reduction in infarct size was observed. Although administration of this ester appeared safe and had protective properties in this model, it also is not commercially available at this time.
CYSTEINE, CYSTINE, AND ACETYLCYSTEINE
Cysteine usually is nonessential in the diet because it can be synthesized endogenously from methionine and phenylalanine.39,76 Impaired synthesis of cysteine from methionine may necessitate the provision of a source of cysteine to some patients with cirrhosis; however, supplementation with L-cysteine could lead to hypercysteinemia and potential toxicity.77 Administration of oral L-Cysteine to patients with cirrhosis has been noted to cause a twofold greater maximal plasma cysteine concentration and plasma elimination half-life, and a delayed excretion of metabolic end products when compared with those of controls. However, an impaired cysteine uptake from the plasma has been proposed secondary to a decrease in plasma GSH. Another example of precursor use involves L-2-oxothiazofidine-4-carboxylate, which is converted to S-carboxyl-L-cysteine and undergoes spontaneous decarboxy-lation to liberate L-Cysteine, thereby supporting GSH synthesis.8
In noncirrhotic, malnourished patients receiving parenteral nutrition, deficiencies in cysteine (and other amino acids) also can occur, possibly caused by the loss of the first-pass delivery of methionine to the liver and portal blood flow.31 As mentioned previously, cysteine is oxidized easily into cystine, and therefore, is not easily incorporated (and minimally present) in standard crystalline amino acid solutions used in intravenous hyperalimentation. This has resulted in hypo-cystinemia, with plasma cystine concentrations decreasing to 30% below baseline, as described in 12 patients with cirrhosis who received a FreeAmmine II source of amino acids in their intravenous hyperalimentation.76
NAC is available, as Mucomyst (Apothecon, Division of Bristol-Myers Squibb, Princeton, NJ) and generic products, and has been used for years as an antidote to acetaminophen toxicity. NAC may be a direct source of cysteine following hydrolysis or may reduce plasma cystine through thiol- disulfide exchange, liberating endogenous cysteine.8 Under normal conditions (no GSH deficiency), NAC does not increase total GSH, since the intracellular concentration is under feedback control. Recently, despite administration of NAC 600 mg po tid, a sustained increase in GSH concen-trations could not be found in the plasma, bronchoalveolar lavage fluid, or lung tissue of patients with chronic obstructive pulmonary disease.46
S-ADENOSYL- L-METHIONINE
Hepatic GSH concentrations have been restored to nearly normal in liver biopsies of patients with cirrhosis following long-term oral SAM administration.23 Studies have documented improvement in pruritus, jaundice, and bio-chemical parameters in patients with intrahepatic cholestasis of pregnancy treated with SAM.33 The clinical efficacy of SAM in the treatment of cholestasis associated with hepatic diseases has been reviewed.78 For the treatment of liver disorders, such as intrahepatic cholestasis, the recommended dose of SAM is 800 mg parenterally or 1600 mg/d orally. 23,78
Loguercio et al.78 administered SAM 2 g/d iv for a total of 15 days to 20 patients with biopsy-proven alcoholic cirrhosis. An increase in red blood cell GSH (to 2.20 ± 1.10 mM/L from a baseline of 1.60 ± 0.97 mM/L) and a decrease in cysteine content (to 65 ± 14 M/L from 122 ± 42 M/L) was demonstrated.
The cysteine groups of SAM synthetase might be protected from oxidation by a normal concentration of GSH.80 When there is a reduction in liver GSH or increased concentrations of GSSG by toxin or disease, a vicious cycle might start. Depletion of GSH could lead to inactivation of SAM synthetase, with further decrease in GSH concentrations. worsening the deficiency in SAM synthetase. In this context, SAM administration may act as a precursor for GSH synthesis and also bypass the deficiency in SAM synthetase. SAM is not commercially available in the US.
Summary
The importance of GSH in health and disease is a subject of active research, presentation, and publication. GSH appears to be metabolically important in a wide variety of disease states, only a few of which have been discussed. At our institution, GSH has been the topic of grand rounds, and we have attempted to modify GSH stores in long-term parenteral nutrition patients with cirrhosis. We believe the significance and widespread use of GSH will only increase with time, and have attempted to introduce the topic to a broad range of clinicians. As pharmaceutical manipulation of GSH concen-trations is an intervention likely to increase within the coming years, health clinicians need to be aware of the rationale of such attempts and methods of administering bioavailable forms of GSH or its substrates.
References
| 1. | Meister A. Glutathione metabolism and its selective modification. J Biol Chem 1988;263:17205-8. |
| 2. | Altomare E, Vendemiale G, Alano O. Hepatic glutathione content in patients with alcoholic and non alcoholic liver diseases. Life Sci 1988;43: 991-8. |
| 3. | Deneke SM, Fanburg BL. Regulation of cellular glutathione. Lung Cell Mol Physiol 1989;1:LI63-LI73. |
| 4. | Wu D, Meydam SN, Sastre J, Hayek M, Meydani M. In vitro glutathione supplementation enhances interleukin-2 production and mitogenic response of peripheral blood mononuclear cells from young and old subjects. J Nutr 1994;124:655-63. |
| 5. | Anderson ME, Powrie F, Puri RN, Meister A. Glutathione monoethyl ester: preparation, uptake by tissues, and conversion to glutathione. Arch Biochem Biophys 1985;239:538-48. |
| 6. | Meister A, Anderson ME. Glutathione. Ann Rev Biochem 1983;52:711 60. |
| 7. | Flagg EW, Coates RJ, Jones DP, Byers TE, Greenberg RS, Gridley G, et al. Dietary glutathione intake and the risk of oral and pharyngeal cancer. Am J Epidentiol 1994; 139:453-65. |
| 8. | DeLeve LD, Kaplowitz N. Glutathione metabolism and its role in hepatotoxicity. Pharmacol Ther 1991;52:287-305. |
| 9. | Halliwell B. Free radicals, antioxidants, and human disease: curiosity, cause, or consequence? Lancet 1994;344:721-4. |
| 10. | Aw TY, Wierzbicka G, Jones DP. Oral glutathione increases tissue glutathione in vivo. Chem Biol Interact 1991;80:89-97. |
| 11. | Bellomo G, Offhenius S. Altered thiol and calcium homeostasis in oxidative hepatocellular injury. Hepatology 1985;5:876-82. |
| 12. | Toborek M, Hennig B. Fatty acid-mediated effects on the glutathione redox cycle in cultured endothelial cells. Am J Clin Nutr 1994;59:60-5. |
| 13. | Hagen TM, Wierzbicka GT, Sillau AH, Bowman BB, Jones DP. Bioavailability of dietary glutathione: effect on plasma concentration. Am J Physiol 1990;259:524-9. |
| 14. | Hagen TM, Jones DP. Role of glutathione transport in extrahepatic detoxification. In: Sakamoto Y, Higashi T, Taniguchi N, et al., eds. Glutathione centennial: molecular perspectives and clinical implications. New York: Academic Press, 1989:423-33. |
| 15. | Hunjan MK, Evered DF. Absorption of glutathione from the gastrointestinal tract. Biochim Biophys Acta 1985;815:184-8. |
| 16. | Amano J, Suzuki A, Sunamori M. Salutary effect of reduced glutathione on renal function in coronary artery bypass operation. J Am Coll Surg 1994; 179:714-20. |
| 17. | Holroyd KJ, Buhl R, Borok Z, Roum JH, Bokser AD, Grimes GJ, et al. Correction of glutathione deficiency in the lower respiratory tract of H1V seropositive individuals by glutathione aerosol treatment. Thorax 1993;48:985-9. |
| 18. | Kaplowitz N, Aw TY, Ookhtens M. The regulation of hepatic glutathione. Ann Rev Pharmacol Toxicol 1985~25:715-44. |
| 19. | Shimizu M, Morita S. Effects of feeding and fasting on hepatolobular distribution of glutathione and cadmium-induced hepatotoxicity. Toxicology 1992;75:97-107. |
| 20. | Martensson J, Meister A. Mitochondrial damage in muscle occurs after marked depletion of glutathione and is prevented by giving glutathione monoester. Proc Nad Acad Sci U S A 1989;86:471-5. |
| 21. | Griffith OW, Meister A. Glutathione: interorgan translocation, turnover, and metabolisrn. Proc Nad Acad Sci U S A 1979;76:5606- 10. |
| 22. | Chawla RK, Lewis FW, Kutner MH, Bate DM, Roy RGB, Rudman D. Plasma cysteine, cystine, and glutathione. Gastroenterology 1984;87: 770-6. |
| 23. | Friedel HA, Goa KL, Benfield P. S-adenosyl-L-methionine: a review of its pharmacological properties and therapeutic potential in liver dysfunction and affective disorders in relation to its physiological role in cell metabolism. Drugs 1989;38:389-416. |
| 24. | Jochnnann C, Klee S, Ungemach FR, Younes M. The role of glutathione and protein thiols in CBrC13(-) induced cytotoxicity in isolated rat hepatocytes. Pharmacol Toxicol 1994;75:7-16. |
| 25. | Ito Y, Suzuki Y, Ogonuki H, Hiraishi H, Razandi M, Terano A, et al. Role of iron and glutathione redox cycle in apap-induced cytotoxicity to cultured rat hepatocytes. Dig Dis Sci 1994;39:1257-64. |
| 26. | Walsh SW, Wang Y. Deficient glutathione peroxidase activity in preeclampsia is associated with increased placental production of thromboxane and lipid peroxides. Am J Obstet Gynecol 1993; 169: 1456-61. |
| 27. | Sheiner P, De Majo W, Levy GA. Acetylcysteine and fulminant hepatic failure. Hepatology 1992;15:552-4. |
| 28. | Whitcomb DC, Block GD. Association of apap, hepatotoxicity with fasting and ethanol use. JAMA 1994;272:1845-50. |
| 29. | Boesgaarg S, Iversen HK, Wroblewski H, Poulsen HE, Frandsen H, Kastrup J, et al. Altered peripheral vasodilator profile of nitroglycerin during long-term infusion of N-acetylcysteine. J Am Coll Cardiol 1994;23:163-9. |
| 30. | Gilbert HF. Biological disulfides: the third messenger? J Biol Chem 1982;257:12086-9 1. |
| 31. | Corrales F, Cabrero C, Pajares MA, Ortiz P, Martin-Duce A, Mato JM. Inactivation and dissociation of S-adenosylmethionine synthetase by modification of sulfhydryl groups and its possible occurrence in cirrhosis. Hepatology 1990; 11:216-22. |
| 32. | Chawla RK, Bonkovsky HL, Galambos JT. Biochemistry and pharmacology of S-adenosyl-L-methionine and rationale for its use in liver disease. Drugs 1990;40(suppl 3):98-110. |
| 33. | Kaye GL, Blake JC, Burroughs AK. Metabolism of exogenous S-adenosyl-L-methionine in patients with liver disease. Drugs 1990;40 (suppl 3):124-8. |
| 34. | Marchesini G, Bugianesi E, Bianchi G, Fabbri A, Marchi E, Zoli M, et al. Effect of S-adenosyl-L-methionine administration on plasma levels of sulphur-containing amino acids in patients with liver cirrhosis. Clin Nutr 1992; 11:303-8. |
| 35. | Cabrero C, Martin-Duce A, Ortiz P, Alemany S, Mato JM. Specific loss of the high-molecular weight form of S-adenosyl-L-methionine synthetase in human liver cirrhosis. Hepatology 19W8:15304. |
| 36. | Loguercio C, Delvecchio Blanco C, Coltorti M, Nardi G. Alteration of erythrocyte glutathione, cysteine and glutathione synthetase in alcoholic and non-alcoholic cirrhosis. Scand J Clin Lab Invest 1992;52: 207-13. |
| 37. | Shigesawa, T, Sato C, Marurno F. Significance of plasma glutathione determination in patients with alcoholic and non-alcoholic liver disease. J Gasumviterol Hepatol 1992;7:7-11. |
| 38. | Seifert CF, Anderson DC, Bui B, Glore SR, Vndracek TG, Raymond T. Correlation of acetarninophen and ethanol use, plasma glutathione concentrations and diet with hepatotoxicity. Pharmacotherapy 1994; 14: 376-7. |
| 39. | Roederer M, Staal FIT, Ela SW, Herzenberg LA, Herzenberg LA. N- acetylcysteine: potential for AIDS therapy. Pharmacology 1993;46: 121-9. |
| 40. | Roederer M, Ela SW, Staal FJT, Herzenberg LA, Herzenberg LA. N- acetylcysteine: a new approach to anti-HIV therapy. AIDS Res Hum Retroviruses 1992;8:209-17. |
| 41. | Staal FIT, Roederer M, Israelski DM, Bubp J, Mole LA, McShane D, et al. AIDS Res Hum Retroviruses 1992;8:305-11. |
| 42. | Roederer M, Raju PA, Staal FJT, Herzenberg LA, Herzenberg LA. N-acetylcysteine inhibits latent HIV expression in chronically infected cells. AIDS Res Hum Retroviruses 1991;7:563-7. |
| 43. | Roederer M, Staal FIT, Anderson M, Rabin R, Raju PA, Herzenberg LA, et al. Disregulation of leukocyte glutathione in AIDS. Ann N Y Acad Sci 1993;667:113-25. |
| 44. | Staal FIT, Roederer M, Raju PA, Anderson MT, Ela SW, Herzenberg LA, et al. Antioxidants inhibit stimulation of H1V transcription. AIDS Res Hum Retrovirus 1993;9:299-306. |
| 45. | Buhl R, Holroyd KJ, Mastrarigeli A, Cantin AM, Jaffe HA, Wells FB, et al. Systemic glutathione deficiency in symptom free HIV seropositive individuals. Lancet 1989;2:1294-8. |
| 46. | Bridgeman MME, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma, bronchoalveolar lavage fluid and lung tissue. Thorax 1994;49:670-5. |
| 47. | Suter PM, Domenighetti G, Schaller MD, Uverriere MC, Ritz R, Perret C. N-acetylcysteine enhances recovery from acute lung injury in man. Chest 1994;105:190-4. |
| 48. | Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione in patients with sepsis and the adult respiratory distress syndrome. Chest 199 1; 100: 1397-403. |
| 49. | Bunnell E, Pacht ER. Oxidized glutathione is increased in alveolar fluid of patients with adult respiratory distress syndrome. Am Rev Respir Dis 1993;148:1174-8. |
| 50. | Grigg J, Barber A, Silverman M. Bronchoalveolar lavage fluid glutathione in intubated premature infants. Arch Dis Child 1993;69:49-51. |
| 51. | Smith LJ, Houston M, Anderson J. Increased levels of glutathione in bronchoalveolar lavage from patients with asthma. Am Rev RespiR Dis 1993; 147:1461-4. |
| 52. | Johnson ME, Sill JC, Uhl CB, Van Dyke RA. Effect of halothane on hypoxic toxicity and glutathione status in cultured rat hepatocytes. Anesthesiology 1993-,79:1061-71. |
| 53. | Picone TA, Daniels TA, Ponto KH, Pitard WB. Cord blood tryptophan concentrations and total cysteine concentrations. JPEN J Parenter Enteral Nutr 1989; 13:106-7. |
| 54. | Liu JZ, Zhang BZ, Milner JA. Dietary selenite modifies glutathione metabolism and 7,12-dimethylbenz(a)anthracene conjugation in rats. J Nutr 1994;124:172-80. |
| 55. | Fahr MJ, Kombluth J, Blossom S, Schaeffer R, Klinberg VS. Glutamine enhances immunoregulation of tumor growth. JPEN J Parenter Enteral Nutr 1994;18:471-6. |
| 56. | Ahn H, Lee E, Kim K, Lee C. Effect of glutathione and its related enzymes on chemosensitivity of renal cell carcinoma and bladder carcinoma cell lines. J Urol 1993; 151:263-7. |
| 57. | Sian J, Dexter DT, Less AJ, Daniel S, Jenner P, Marsden CD. Glutathione-related enzymes in brain in Parkinson's disease. Ann Neurol 1994;36:356-61. |
| 58. | Jenner P. Oxidative damage in neurodegenerative disease. Lancet 1994;344:796-8. |
| 59. | Henderson A, Hayes P. Acetylcysteine as a cytoprotective antioxidant in patients with severe sepsis: potential new use for an old drug. Ann PharmacoLher 1994;28:1086-8. |
| 60. | Spies CD, Reinhart K, Witt 1, Meier-Hellmann A, Hanneman L, Bredle DL, et al. Influence of N-acetylcysteine on indirect indicators of tissue oxygenation in septic shock patients: results from a prospective, randomized, double-blind study. Crit Care Med 1994;22:1738-46. |
| 61. | Hoshida S, Kuzuya T, Yamashita N, Nishida M, Kitahara S, Hori M,.et al. Gamma-glutamylcysteine ethyl ester for myocardial protein in dogs during ischemic and reperfusion. J Am Coll Cardiol 1994;24: 1391-7. |
| 62. | Blaustein A, Deneke SM, Stolz RI, Baxter D, Healey N, Fanburg BL. Myocardial glutathione depletion impairs recovery after short periods of ischemia. Circulation 1989;80:1449-57. |
| 63. | Singh A, Lee KJ, Lee CY, Goldfarb RD, Tsan MF. Relation between myocardial glutathione content and extent of ischemia-reperfusion injury. Circulation 1989;80:1795-804. |
| 64. | Forman MB, Puett DW, Cates CU, McCroskey DE, Beckman JK, Greene HL, et al. Glutathione redox pathway and reperfusion injury. Circulation 1988;78:202-13. |
| 65. | Duruibe V, Okonmah A, Blyden GT. Effect of cyclosporin on rat liver and kidney glutathione content. Pharmacology 1989;39:205-12. |
| 66. | Inselmann G, Hannemann 1, Baumann K. Cyclosporine A induced lipid peroxidation and influence on glucose-6-phosphate in rat hepatic and renal microsomes. Res Commun Chem Pathol Pharmacol 1990;68: 189-203. |
| 67. | Wang C, Salahudeen AK. Cyclosporine nephrotoxicity: attenuation by an antioxidant-inhibitor of lipid peroxidation in vitro and in vivo. Transplantation 1994;58:940-6. |
| 68. | Julius M, Lang CA, Gleiberman L, Harburg E, DiFranceisco W, Schork A. Glutathione and morbidity in a community-based sample of elderly. J Clin Epiderniol 1994;47:1021-6. |
| 69. | Lang CA, Naryshkin S, Schneider DL, Mills B1, Lindeman RD. Low blood glutathione in healthy aging adults. J Lab Clin Med 1992; 120: 720- 5. |
| 70. | Fletcher RH, Fletcher SW. Glutathione and ageing: ideas and evidence (editorial). Lancet 1994;344:1379-80. |
| 71. | Jones DP, Coates RJ, Flagg EW, Eley JW, Block G, Greenberg RS, et al. Glutathione in foods listed in the National Cancer Institute's health habits and history food frequency questionnaire. Nutr Cancer 1992; 17: 57-75. |
| 72. | Davis MA, Wallig MA, Eaton D, Borroz 1, Jeffery EH. Differential effect of cyanohydroxybutene on glutathione synthesis in liver and pancreas of male rats. Toxicol Appl Pharmacol 1993; 123:257-64. |
| 73. | Jones DP, Hagen TM, Weber R, Wierzbicka GT, Bonkovsky HL. Oral administration of glutathione (GSH) increases plasma GSH concentrations in humans (abstract). FASEB J 1989;3:A 1250. |
| 74. | Witschi A, Reddy S, Stofer B, Lauterburg BH. The systemic availability of oral glutathione. Eur J Clin Pharmacol 1992;43:667-9. |
| 75. | Cook GC, Sherlock S. Results of a controlled clinical trial of glutathione in cases of hepatic cirrhosis. Gut 1965;6:472-6. |
| 76. | Rudman D, Kutner M, Ansley J, Jansen R. Chipponi J, Bain RP. Hypotyrosinemia, hypocystinernia, and failure to retain nitrogen during total parenteral nutrition in cirrhotic patients. Gastroenterology 198 1;8 1: 1025-35. |
| 77. | Tribble DL, Jones DP, Ardehali A, Feeley RM, Rudman D. Hypercysteinemia and delayed sulfur excretion in cirrhotics after oral cysteine loads. Am J Clin Nutr 1989;50:1401-6. |
| 78. | Almasio P, Bortolini M, Pagliaro L, Coltorti M. Role of S-adenosyl- methionine in the treatment of intrahepatic cholestasis. Drugs 1990;40 (suppI 3):111-23. |
| 79. | Loguercio C, Del Vecchio C, Coltorti M. Effects of intravenous S- adenosyl-L-methionine on red blood cell content of glutathionine and cyteine in alcoholic cirrhosis (abstract). Gastroenterology 1993;104: A944. |
| 80. | Ortiz P, Moreno J, Puerta JL, Mato JM. S-adenosyl-L-methionine and the liver. Ital J Gastroenterol 1993;25:135-7. |